Projects
Work done and those in progress.
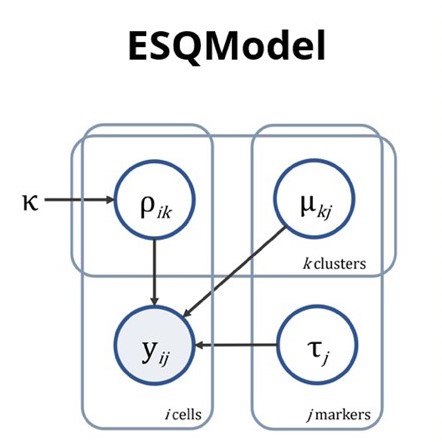
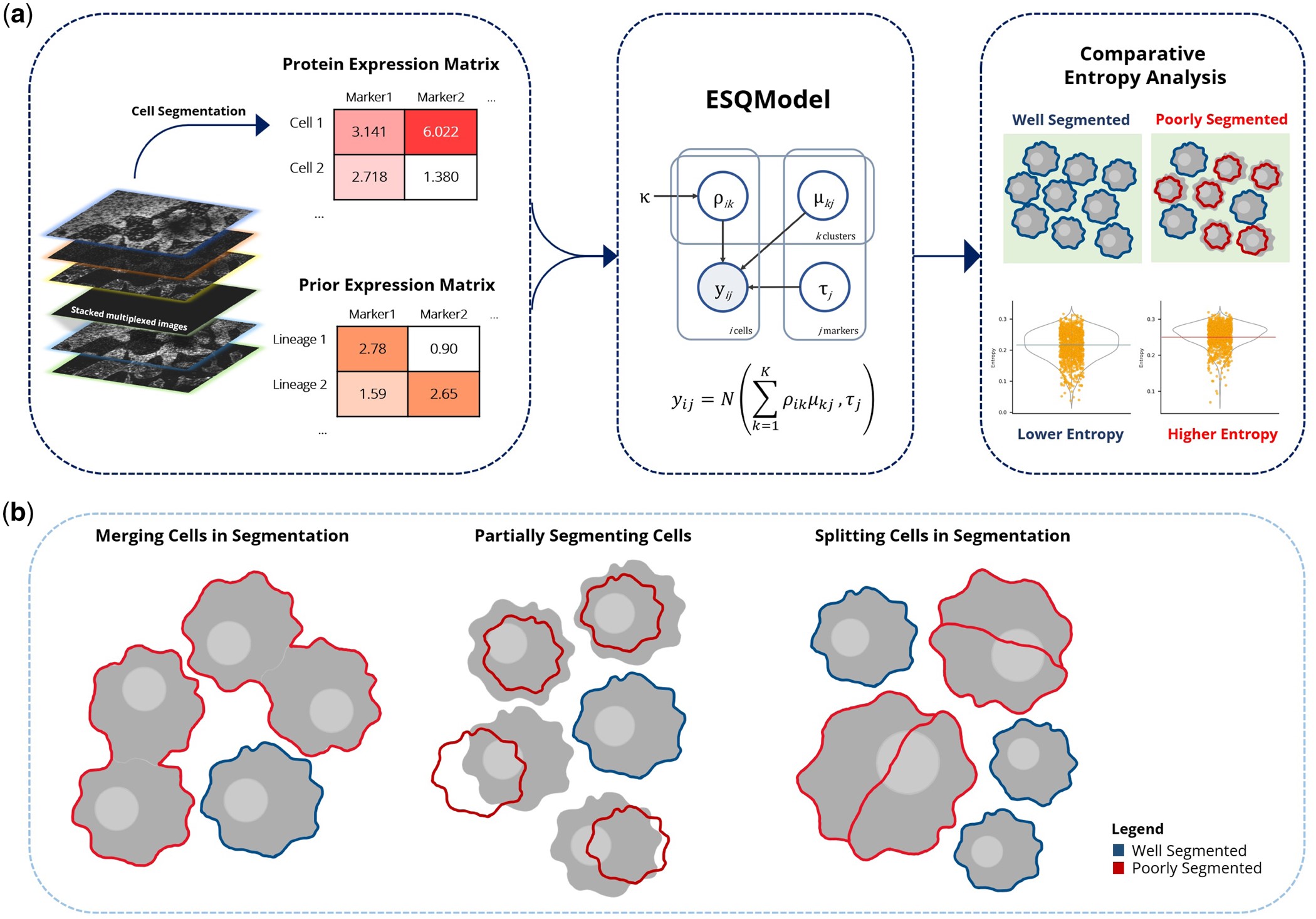
ESQmodel: biologically informed evaluation of 2-D cell segmentation quality in multiplexed tissue images
Lee et al. 2024, Bioinformatics
Abstract
Motivation Single cell segmentation is critical in the processing of spatial omics data to accurately perform cell type identification and analyze spatial expression patterns. Segmentation methods often rely on semi-supervised annotation or labeled training data which are highly dependent on user expertise. To ensure the quality of segmentation, current evaluation strategies quantify accuracy by assessing cellular masks or through iterative inspection by pathologists. While these strategies each address either the statistical or biological aspects of segmentation, there lacks a unified approach to evaluating segmentation accuracy.
Results In this article, we present ESQmodel, a Bayesian probabilistic method to evaluate single cell segmentation using expression data. By using the extracted cellular data from segmentation and a prior belief of cellular composition as input, ESQmodel computes per cell entropy to assess segmentation quality by how consistent cellular expression profiles match with cell type expectations.
Available on Github!

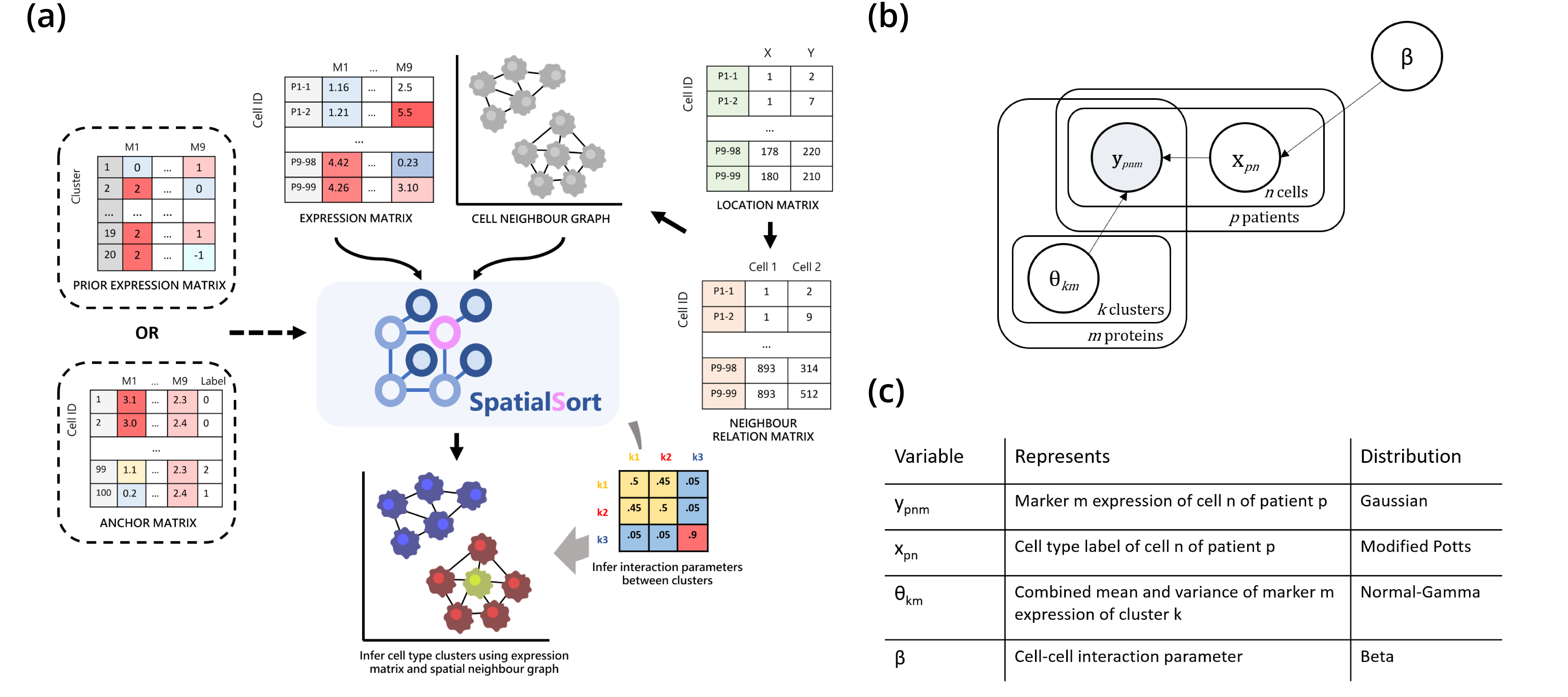
SpatialSort: a Bayesian model for clustering and cell population annotation of spatial proteomics data
Lee et al. 2023, Bioinformatics
Abstract
Motivation Recent advances in spatial proteomics technologies have enabled the profiling of dozens of proteins in thousands of single cells in situ. This has created the opportunity to move beyond quantifying the composition of cell types in tissue, and instead probe the spatial relationships between cells. However, most current methods for clustering data from these assays only consider the expression values of cells and ignore the spatial context. Furthermore, existing approaches do not account for prior information about the expected cell populations in a sample.
Results To address these shortcomings, we developed SpatialSort, a spatially aware Bayesian clustering approach that allows for the incorporation of prior biological knowledge. Our method is able to account for the affinities of cells of different types to neighbour in space, and by incorporating prior information about expected cell populations, it is able to simultaneously improve clustering accuracy and perform automated annotation of clusters. Using synthetic and real data, we show that by using spatial and prior information SpatialSort improves clustering accuracy. We also demonstrate how SpatialSort can perform label transfer between spatial and nonspatial modalities through the analysis of a real world diffuse large B-cell lymphoma dataset.
Available on Github!
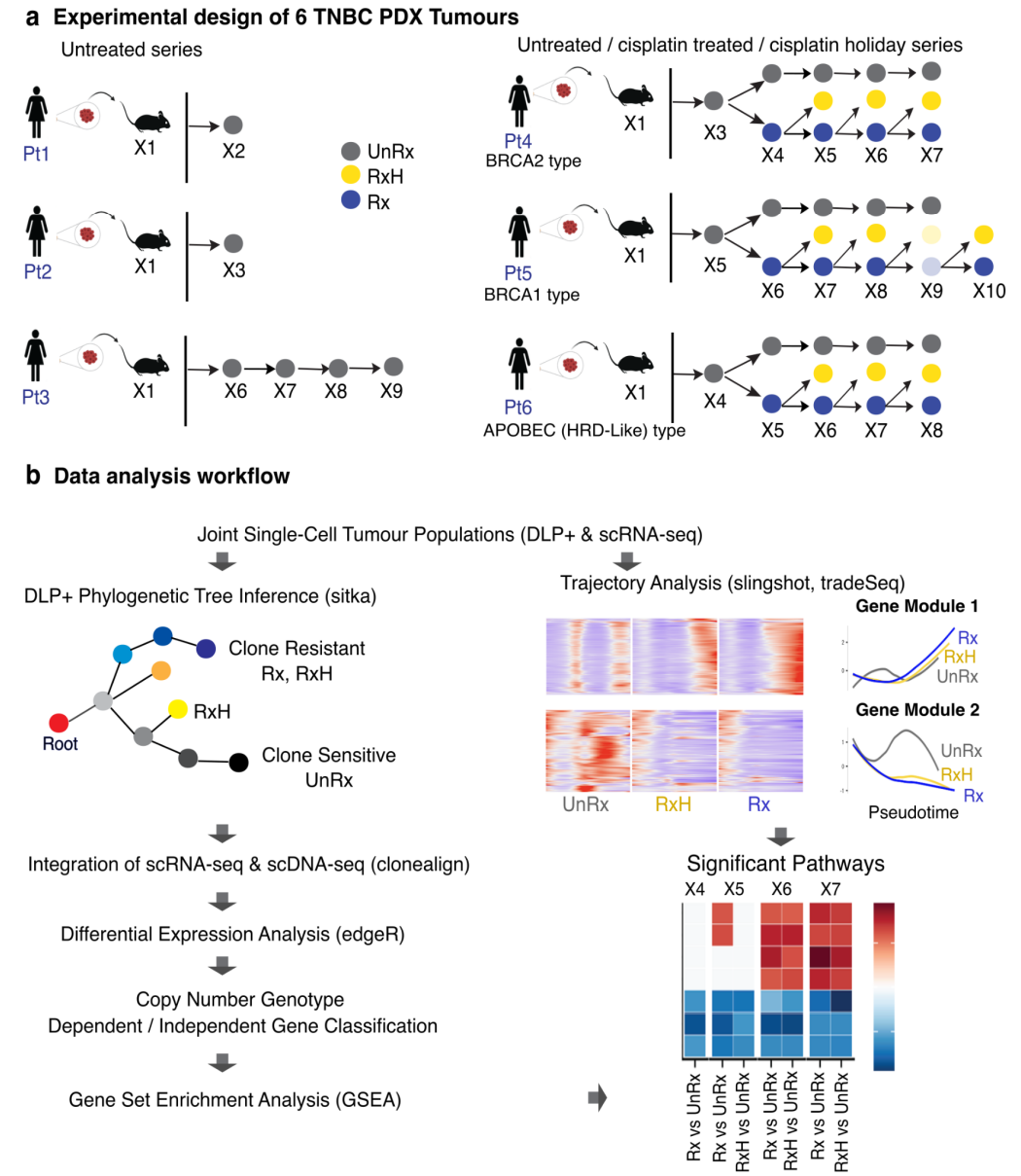
Single cell decoding of drug induced transcriptomic reprogramming in triple negative breast cancers
Kabeer, Tran and Andronescu et al. 2023, bioRxiv
Abstract
Background The encoding of cell intrinsic resistance states in breast cancer reflects the contributions of genomic and non-genomic variation. However, identifying the potential contributions of each requires accurate measurement and subtraction of the contribution of clonal fitness from co-measurement of transcriptional states. Somatic genomic variation in gene dosage, copy number variation, is the dominant mutational mechanism in breast cancer contributing to transcriptional variation and has recently been shown to contribute to platinum chemotherapy resistance states. Here we deploy time series measurements of triple negative breast cancer single cell transcriptomes in conjunction with co-measured single cell copy number associated clonal fitness to identify the contributions of genomic and non-genomic mechanisms to drug associated transcription states.
Results We generated serial scRNA-seq data (126,556 cells) from triple negative breast cancer (TNBC) patient-derived xenograft (PDX) experiments over 2.5 years in duration, and matched it against genomic copy number single cell data from the same biological samples. We show that the cell memory of transcriptional states of TNBC tumors serially exposed to platinum identifies distinct clonal responses within individual tumours. Copy-number clones with high drug fitness leading to clonal sweeps exhibit less transcriptional reversion, whereas clones with weak drug fitness exhibit highly dynamic transcription on drug withdrawal. Pathway analysis shows that copy number associated and copy number independent transcripts converge on epithelial-mesenchymal transition (EMT) and cytokine signaling states associated with resistance. We show from trajectory analysis that transcriptional reversion exhibits hysteresis, indicating that new intermediate transcriptional states are generated by platinum exposure.
Conclusions We discovered that copy number clones with strong genotype associated fitness under platinum became fixed in their states, resulting in minimal transcriptional reversion on drug withdrawal. In contrast clones with weaker fitness undergo non-genomic transcriptional plasticity and these distinct responses co-exist within single tumours. Together the data suggest that copy number associated and copy number independent transcriptional states may contribute to platinum drug resistance within individual tumours. The dominance of genomic or non-genomic mechanisms within individual polyclonal tumours has implications for approaches to restoration of drug sensitivity and re-treatment strategies.
Available on Github!